What are Caspases?
Caspase Structure Determines Function
The currently known mammalian caspases may be categorized into three groups: apoptosis initiators, apoptosis executioners, and inflammatory caspases. Caspases -2, -8, -9, and -10 are apoptosis initiators and the apoptosis executioners are caspases -3, -6, and -7 (McIlwain et al, 2013). Human caspases -1, -4, -5, and -12-L have been implicated not in apoptosis but in "innate immune responses and the regulation of inflammatory cytokine processing" (Taylor et al, 2008). Interestingly, caspase-12 "is expressed as a truncated, catalytically inactive protein in most humans," known as caspase-12-S (Taylor et al, 2008). However, full-length caspase-12-L is present in some individuals of African descent, who "appear to be more susceptible to inflammatory diseases" (Taylor et al, 2008). Somewhat of an outlier, caspase-14 does not fit into any of the main caspase categories, but is involved in cornification. Cornification refers to the remodeling event characteristic of the late stage of keratinocyte differentiation, which leads to the formation of the water-impermeable barrier of the skin (Crawford et al, 2011). Hence, caspase-14 is expressed in the epidermis (Shalini et al, 2015).

Figure 1. (McIlwain et al, 2013). Domain structure of the known human caspases in zymogen form. Every caspase contains a large (orange) and small (red) subunit, with the exception of caspase 12-S. All apoptotic executioner caspases (-3, -6, and -7) are composed of the small and large subunits only. Initiator caspases-9 and -2 as well as inflammatory (non-apoptotic) caspases-1, -4, -5, and 12-L contain the DED (death effector domain) at the N-terminus (green). Two copies of the N-terminal prodomain CARD (caspase activation and recruitment domain; blue) are present in initiator caspases-8 and -10.
Caspase activation is usually initiated through proteolytic processing of the caspase between the large and small subunits, which form a heterodimer (Tait and Green, 2010, Figure 2). During the processing event, the caspase is rearranged into an active conformation (Taylor et al, 2008). In the active conformation, the active site cysteine is exposed and can interact with an aspartate residue on the substrate, resulting in substrate cleavage. In healthy, non-apoptotic cells, initiator caspases exist as monomers with prodomains, whereas effector/executioner caspases are present as pre-formed dimers consisting of only the large and small subunits (Taylor et al, 2008). After modification by an initiator caspase, the effector caspase dimers undergo a subsequent dimerization, resulting in active heterotetramers (Taylor et al, 2008). Earnshaw et al in 1999 provides a more extensive discussion of the biochemistry behind caspase catalysis.

Figure 2 (Tait and Green, 2010). A) Initiator procaspases (zymogens)-2, -8, and -9 consist of an N-terminal prodomain, large subunit and small subunit. Executioner procaspases-3, -6, and -7 consist of linked large and small subunits. B) Activation of initiator caspases such as caspase-8 involves interaction with a death domain ligand, dimerization, and cleavage of the prodomain and between the large and small subunits. Executioner caspase activation is completed with cleavage between the large and small subunits.
Caspase Involvement in Extrinsic and Intrinsic Apoptosis
The cascade of caspase activation depends on the apoptotic pathway being activated (McIlwain et al, 2013; Figure 3). In the extrinsic pathway, a ligand such as Fas ligand or TNF-α binds to a death receptor at the cell membrane, leading to the formation of a death-inducing signaling complex (DISC). Caspase-8 is activated by association with the DISC and goes on to cleave (activate) effector caspases -3 and -7 directly and/or to cleave Bid, which leads to mitochondrial permeabilization by Bax and Bak (Crawford et al, 2011). In the intrinsic pathway, cellular damage signals act through p53 and other sensors to activate the BH3-only proteins, which antagonize the Bcl-2 family of apoptotic inhibitors, leading to permeabilization of the outer mitochondria by Bax and Bak. The two pathways converge on mitochondrial permeabilization and the release of cytochrome c (Cyt. C) to the cytosol. Cytochrome c, Apaf1, and procaspase-9 form a complex known as the apoptosome, the scaffold for activating caspase-9. Smac/DIABLO is also released from mitochondria and antagonizes the IAPs (inhibitor of apoptosis proteins), which normally inhibit caspase activity. Caspase-9 goes on to activate the effector caspases, which cleave numerous substrates and execute cell death (Crawford et al, 2011). The apoptotic initiator and effector caspases mediate the morphological changes involved with apoptosis. In both the intrinsic and extrinsic apoptotic pathways, initiator caspases act as signal transducers, directing the cell toward death by cleaving a small number of substrates. Substrates cleaved by the initiator caspases -2, -8, -9 and -10 include effector caspases -3, -6, and -7 (which are activated), and inhibitors of apoptosis (which are inactivated) (Tait and Green, 2010). The intrinsic and extrinsic pathways converge with activation of the effector caspases, which kill the cell by cleaving a wide range of substrates. Both the extrinsic and intrinsic apoptotic pathways result in a cell exhibiting the characteristic apoptotic morphology of blebbing, DNA fragmentation, and loss of mitochondrial membrane potential. Apoptotic physical changes are largely mediated by downstream cleavages of caspase substrates (Crawford et al, 2011). Consult the interactive Apoptosis Signaling Pathway by R&D Systems to view a more complex portrayal of caspase involvement in the intrinsic and extrinsic pathways of apoptosis. Take note of the number of pathways converging to or diverging from Caspases 8 and 10, the initiator caspases of the extrinsic apoptotic pathway.

Figure 3 (McIlwain et al, 2013). Caspase activation cascades in extrinsic and intrinsic apoptosis. Extrinsic apoptosis is stimulated by ligand binding to death receptors and depends on caspase-8 activation. Intrinsic apoptosis is stimulated by stressors such as DNA damage, ROS, and unfolded protein accumulation, and is dependent on caspase-9 activation at the apoptosome. Common to each apoptotic pathway is the activation of caspases-3, -6, and -7 and subsequent cleavage of cellular substrates by effector caspases.
Caspase Substrates
- Apoptosis regulation (Apaf-1, Bad, Bax, Bcl-2 Bcl-xL, Bid)
- Cell adhesion (β-catenin, plakoglobin, E-cadherin, paxillin)
- Cell cycle regulation (Cdc6, NuMa, Cyclin E, Rb)
- Cytoskeleton formation (β-actin, filamin, plectin, β-spectrin, α-tubilin, troponin T, vimentin)
- DNA binding (CREB, c-Rel, NF-κB p50 and p65 subunits, SREBP-1 and -2, STAT1)
- DNA synthesis, cleavage, and repair (topoisomerase I, ICAD, helicard, MCM3)
- RNA synthesis and splicing (BTF3, nucleolin, RHA)
- Neurodegeneration (APP, ataxin-1, Huntingtin, Parkin)
- Nuclear Structure (Lamins A, B1, and 3, emerin, scaffold attachment factor-A)
- Translation (60S acidic ribosomal P0, eIF3, elF4B,)
- Protein modification (FTase, GGTase I, O-GlcNAse)
- Protein degradation (calpsatain, Cbl, UFD2)
- Calcium, cAMP, cGMP, and lipid metabolism (CCT-α, PDE6, PLC-Ɣ1)
- G-protein signaling (rabaptin-5, Rac, Ras-GAP)

Figure 4 (Martin et al, 2010). Caspase activation in apoptotic and secondary necrotic cells inactivates DAMPS and leads to an anti-inflammatory response and signal macrophages to clean up cellular debris. By contrast, the absence of caspase activation during necrotic cell death allows active DAMPs to exit the cell, resulting in a pro-inflammatory immune response.
Although the 280 caspase substrates reported by Fischer et al in 2003 was striking, over 1500 substrates were known as of 2014 (Kumar et al, 2014). Bioinformatics specialists at Sanford Burnham Medical Research Institute developed CaspDB, a database of known and predicted caspase substrates in the human proteome. CaspDB highlights potential caspase cleavage site sequences in all human proteins and their orthologs and is searchable by protein name, Uniprot ID, and motif (Kumar et al, 2014). Information regarding single nucleotide polymorphisms and post-translational modifications with potential to alter caspase cleavage is included in the database (Kumar et al, 2014).
Presence of a conserved caspase cleavage motif does not guarantee cleavage. (Fischer et al, 2003). Consequently, each predicted caspase cleavage must be experimentally confirmed. Experiments used to elucidate caspase substrates can be divided into two broad categories with regards to the biological question being asked (Crawford et al, 2011; Yun et al in 2014; Figure 5). Forward (in vivo) experiments ask what substrates are cleaved during a biologically relevant apoptotic event. Reverse (in vitro) experiments ask what substrates a particular caspase is capable of cleaving [and at what site(s)]. Although considered in vivo because caspase cleavage occurs in living cells, forward assays are performed on in vitro cell cultures. Both methods must be addressed when validating a functionally relevant cleavage (Crawford et al, 2011). In some cases, a caspase may be capable of cleaving a protein in vitro but may not have access to the same protein in vivo. In other cases, a protein may be known to be cleaved during apoptosis, but an in vitro assay can help clarify which caspase or other protease is responsible for the cleavage. Yun et al in 2014 used an in vitro assay to identify anamorsin as a substrate of caspase-3 in neuronal cells. Full-length anamorsin was radiometrically labeled and incubated with caspase-2, -3, -6, -7, -8, or -9. Cleaved (25-kDa) anamorsin was only detected when caspase-3 was present. Cleavage by caspase-3 during apoptosis nullifies the pro-survival function of full-length anamorsin. Furthermore, cleaved anamorsin was also found in post-mortem neural tissue of patients with Alzheimer's and Parkinson's (Yun et al, 2014), pointing to inappropriate caspase activity as a contributing factor in neurodegenerative diseases.
 |
| Figure 5 (Crawford et al, 2011). (a) Schematic representation of forward (in vivo) caspase substrate discovery techniques. Healthy cells are perturbed to induce apoptosis. Cells are lysed, and cleavage products resulting from the perturbation are identified. (b) Schematic representation of reverse (in vitro) caspase substrate discovery techniques. Healthy cells are lysed, and the lysate is incubated with a caspase. Resulting cleavage products are identified through various techniques.
Caspase inhibitors can also be used to confirm cleavage of a substrate of interest. In the Yun et al, 2014 study discussed above, the pan-caspase inhibitor z-VAD-fmk blocked anamorsin cleavage, indicating caspase involvement in the cleavage. However, pan-caspase inhibitors are only useful to determine general caspase involvement and specific caspase inhibitors provide more precision. If a specific caspase inhibitor suppresses cleavage of a protein, the cleavage is considered dependent on the inhibited caspase. One drawback of all caspase substrate studies remains: identified cleavage products may not be the direct result of caspase activity but of downstream proteases activated by caspases (Yun et al, 2014).
|
Caspase Activity Perpetuates Apoptosis
{kind=link}
 |
| Figure 6 (Guerrero et al, 2012).
Promotion of caspase activation by caspase-9-mediated
feedback amplification of mitochondrial damage.
|
{kind=link}
 |
| Figure 7 (Ojha et al, 2015). Effector caspase-mediated cleavage of autophagic proteins. |
Targeting Caspases to Manipulate Apoptosis
Degenerative diseases like Alzheimer's and Parkinson's are characterized by high rates of apoptotic cell death (Elkshyyan and Aw, 2004), while cancerous tissue often exhibits insufficient apoptosis and overexpression of pro-survival proteins (Plati et al, 2011; Letai, 2008). Consequently, researchers are seeking methods to activate or inhibit caspases to provide the desired effects on apoptosis according to the disease in question.
Another example of caspase manipulation for clinical purposes is the use of resveratrol. Resveratrol (3,4′,5-trihydroxy-trans-stilbene) is a polyphenol abundant in red wine, grapes, peanuts and other plant products. Resveratrol is of interest to researchers due to its apparent cytoprotective and anti-carcinogenic effects (Trincheri et al, 2007). Ulakcsai et al in 2015 investigated the efficiency of resveratrol to attenuate
the effects of age-related degenerative diseases. Serum deprived primary mouse
embryonic fibroblasts showed upregulated caspase-3 and increased apoptosis. However,
when the non-transformed fibroblast cells were exposed to resveratrol at doses of
50 µM or higher, caspase-3 activity was significantly reduced and cells were protected
against apoptotic cell death. Seemingly contradictory properties of resveratrol have been reported in the literature. Resveratrol appears to exert antioxidant effects in some circumstances and pro-oxidant effects in others (de La Lastra and Villegas, 2007; Ahmad et al, 2003), and both pro- and anti- apoptotic effects have been observed. Resveratrol downregulates caspases and apoptosis in non-transformed cell lines, but induces caspase activation and apoptotic cell death in malignantly transformed (cancerous) cells (Ulakcsai et al, 2015). For example, Trincheri et al in 2007 found that resveratrol activated the caspase-dependent intrinsic pathway of apoptosis in human colorectal cancer cells. Furthermore, Alkhalaf et al in 2008 observed apoptosis induction in human breast cancer cells exposed to resveratrol. Cleavage of caspase-3 increased with increasing time and concentration of resveratrol treatment (Alkhalaf et al, 2008; Figures 10 and 11). Treatment with 50 μM resveratrol resulted in cleaved PARP, a marker of apoptosis. Caspase-3 inhibitor DEVD-CHO suppressed PARP cleavage, indicating that the apoptosis induction is dependent on caspase-3 (Alkhalaf et al, 2008; Figure 12). Conversely, Zhang et al in 2015 reported that resveratrol "attenuated the overexpression" of apoptosis markers caspase-4, CHOP, and caspase-3 in human bronchial epithelial cells exposed to cigarette smoke. Zhang et al proposed that resveratrol could be used to protect against cigarette smoke-induced apoptosis, one of the pathogenic factors contributing to chronic obstructive pulmonary disease. The opposite effects of resveratrol on transformed and non-transformed cells appears advantageous for treatment of both degenerative diseases and cancer, as the former seeks to prevent apoptosis of healthy cells without tumorigenesis and the latter aims to cause apoptosis in cancer cells without affecting the surrounding tissue.



Several studies support the idea that the anticancer effects of resveratrol extend beyond apoptosis induction and are mediated by AMPK (AMP-activated protein kinase) activation (Kim and He, 2013; Figure 13). Hwang et al in 2007 found that resveratrol treatment activated AMPK, attenuated growth and stimulated apoptosis in HT-29 colon cancer cells. Interestingly, the cells susceptible to resveratrol had previously been determined resistant to the chemotherapy drug etoposide (Hwang et al, 2007). Reported cancer treatment pathways involving AMPK also include induction of autophagic cell death in chronic myeloid leukemia cells (Puissant et al, 2010), proliferation inhibition of breast cancer cells (El-Masry et al, 2012), and sensitization of glioblastoma multiforme cells to the chemotherapy drug temozolamide (Lin et al, 2010). AMPK activation by resveratrol also resulted in increased sensitivity to ionizing radiation treatment in lung, prostate, and breast cancer cells in vitro (Sanli et al, 2010). The involvement of AMPK in each pathway led to the emergence of AMPK as a target for upregulation in cancer treatment (Kim and He, 2013).

As the main effectors of apoptosis, caspases are popular targets for upregulation by chemotherapeutic agents. For example, the taxane docetaxel was shown to induce apoptosis in a panel of human melanoma cell lines in a caspase-2 dependent manner (Mhaidat et al, 2007). Addition of pan-caspase inhibitor z-VAD-fmk suppressed the docetaxel-induced cell death by 67%, and caspase-2 inhibitor z-VDVAD-fmk resulted in 56% suppression (Mhaidat et al, 2007). Furthermore, an assay measuring caspase activity over time revealed caspase-2 as the first to be activated. (Mhaidat et al, 2007; Figure 8).

Conventional chemotherapeutic agents such as docetaxel and trabectedin above slow cancer progression by causing malignant cells to undergo apoptosis (Mhaidat et al, 2007, Acikgoz et al, 2015; Preusser et al, 2015). However, the development of drug resistance is considered to be the major cause for the failure of chemotherapy in many types of cancer. One of the mechanisms by which tumor cells become resistant to chemotherapy is through the acquired ability to avoid cell death by the upregulation of proteins that inhibit apoptosis and promote cell survival. When the majority of cancer cells are killed by a drug, a few cells with high expression of apoptosis inhibitors are left behind and become the predominant population. Apoptosis-resistant cells eventually proliferate and are responsible for cancer recurrence post-chemotherapy. To target cancer cells resistant to conventional chemotherapy, some researchers are working to develop vaccines against cells expressing high levels anti-apoptotic molecules. Andersen et al in 2005 write, "anti-apoptotic molecules that enhance the survival of cancer cells and facilitate their escape from cytotoxic therapies represent prime candidates as vaccination antigens." If cancer cells not killed by drugs could be targeted by vaccination-induced T cells, the synergy of chemotherapy and vaccination may provide a more effective treatment than either treatment alone (Andersen et al, 2005; Figure 9).
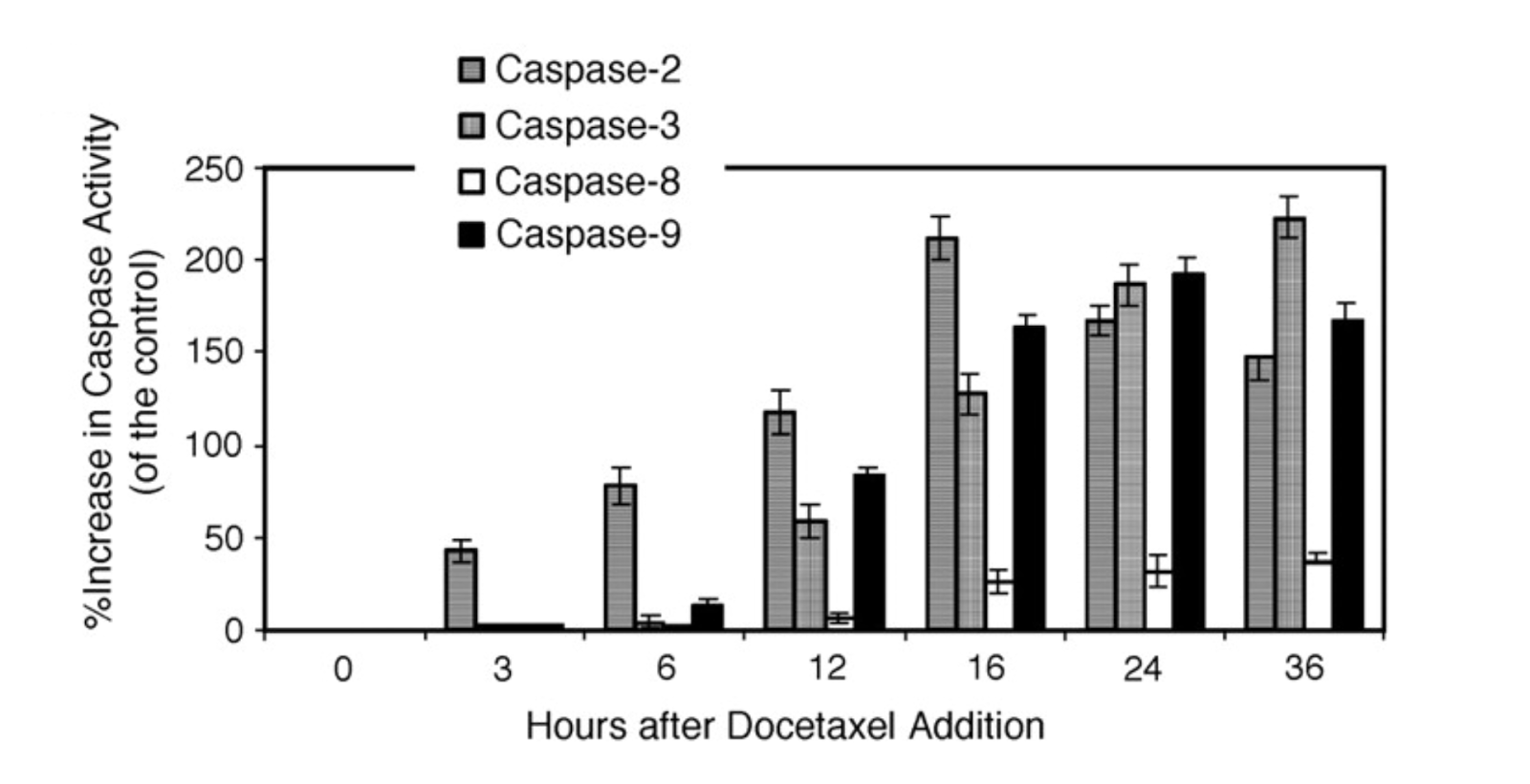
Figure 8 (Mhaidat et al, 2007). Docetaxel induces a cascade of caspase activation in a human melanoma cell line.
Caspase-2 activity precedes activity of the other initiator caspases -8 and -9 and is responsible for activating the effector caspase-3. Method: IgR3 (human melanoma) cells were treated with 20 nM docetaxel. At each indicated time point, cell lysate was analyzed for caspase activity using caspase-specific fluorescent AFC substrates. Each column represents the mean of three individual experiments ± standard error.
The chemotherapy drug trabectedin is already used to treat advanced soft tissue sarcoma and ovarian cancer, but may be used to induce caspase-mediated apoptosis in a wider range of cancer types (Preusser et al, 2015). In an in vitro study on human prostate cancer stem cells, trabectedin induced apoptosis in a dose-dependent manner. Cell death was accompanied by increased expression of caspases -3, -8, and -9 and decreased expression of pro-survival molecule bcl-2 (Acikgoz et al, 2015). Meningioma (intracranial tumor) cells also exhibited heightened activation of caspase-3 and -7, and "massive apoptosis induction" in response to trabectedin treatment (Preusser et al, 2015).
Conventional chemotherapeutic agents such as docetaxel and trabectedin above slow cancer progression by causing malignant cells to undergo apoptosis (Mhaidat et al, 2007, Acikgoz et al, 2015; Preusser et al, 2015). However, the development of drug resistance is considered to be the major cause for the failure of chemotherapy in many types of cancer. One of the mechanisms by which tumor cells become resistant to chemotherapy is through the acquired ability to avoid cell death by the upregulation of proteins that inhibit apoptosis and promote cell survival. When the majority of cancer cells are killed by a drug, a few cells with high expression of apoptosis inhibitors are left behind and become the predominant population. Apoptosis-resistant cells eventually proliferate and are responsible for cancer recurrence post-chemotherapy. To target cancer cells resistant to conventional chemotherapy, some researchers are working to develop vaccines against cells expressing high levels anti-apoptotic molecules. Andersen et al in 2005 write, "anti-apoptotic molecules that enhance the survival of cancer cells and facilitate their escape from cytotoxic therapies represent prime candidates as vaccination antigens." If cancer cells not killed by drugs could be targeted by vaccination-induced T cells, the synergy of chemotherapy and vaccination may provide a more effective treatment than either treatment alone (Andersen et al, 2005; Figure 9).
 |
| Figure 9 (Andersen et al, 2005). Therapeutic strategies: co-targeting chemotherapy to induce apoptosis in cancer cells. Chemotherapeutic agents such as docetaxel (Mhaidat et al, 2007) and trabectedin (Preusser et al, 2015; Acikgoz et al, 2015) kill malignant cells susceptible to apoptosis (shown in cream) and delay the onset of uncontrolled disease. However, a small population of cells remain- cells resistant to chemotherapy due to overexpression of regulators (inhibitors) of apoptosis. These cells (shown in grey) proliferate and eventually result in uncontrolled disease and death. Vaccination against cells with high expression of apoptosis inhibitors could potentially and eradicate all cancer cells if combined with chemotherapy. |
Plant-derived Molecules

Figure 10 (Alkhalaf et al, 2008). Resveratrol (RSVL) induces caspase-3 activation (cleavage) in human breast cancer cells in a time-dependent manner. As time (hours) of 50 μM resveratrol treatment increases, caspase-3 is cleaved into 12, 17, and 20 kDa fragments. Western blot representative of 3 independent experiments. Actin (loading control) expression was the same in each group.

Figure 11 (Alkhalaf et al, 2008). Resveratrol (RSVL) induces caspase-3 activation (cleavage) in human breast cancer cells in a concentration-dependent manner. Western blot representative of 3 independent experiments shows a decreasing signal for 35 kDa uncleaved caspase-3 and increasing signals for cleaved caspase-3 fragments as resveratrol concentration increases.

Figure 12 (Alkhalaf et al, 2008). Treatment with 50 μM resveratrol (RSVL) induced PARP cleavage in human breast cancer cells, indicating apoptosis. When 5 μM DEVD-CHO (caspase-3 inhibitor) was added, cleaved PARP was significantly decreased. Taken together, these data support the concept of resveratrol-induced caspase-3 dependent PARP cleavage in human breast cancer cells.

Figure 13 (Kim and He, 2013). Resveratrol exerts anti-cancer effects in a range of tissue types via activation of AMPK (AMP-activated protein kinase).
Phytochemical use in cancer prevention and therapy is not limited to resveratrol and is of interest because phytochemicals are naturally occurring and less harmful than other chemotherapy drugs. In addition to resveratrol, phytochemicals being investigated as anticancer agents include:
- 24-hydroxyursolic acid from persimmon (Khanal et al, 2010)
- catechin from green tea (Park et al, 2009)
- capsaicin from chili pepper (Kim et al, 2007)
- p-HPEA-EDA from olive oil (Khanal et al, 2011)
- curcumin from turmeric (Pan et al, 2008; Lee et al, Jan 2009; Lee et al, Aug 2009)
{kind=link}
No comments:
Post a Comment